1. Introduction
吲哚及其衍生物是一类非常有效的药物中间体。已有不少相关综述报道其合成方法1。我们将一些常用的合成方法简单的列举了出来,供大家在合成此类化合物的时候参考。
1 (a) G. W. Gribble, Contemp.Org. Synth., 1994, 145. (b) U. Pindur and R. Adam, J. Heterocycl.Chem., 1988, 25, 1. (c)C. J. Moody, Synlett, 1994, 681. (d) R. J. Sundberg, Indoles,Academic Press, San Diego, CA, 1996. (e) T. L. Gilchrist, J.Chem. Soc., Perkin Trans. 1, 1999, 2849. (f) G. W. Gribble, J.Chem. Soc., Perkin Trans. 1,2000, 1045.
2. Fischer 吲哚合成
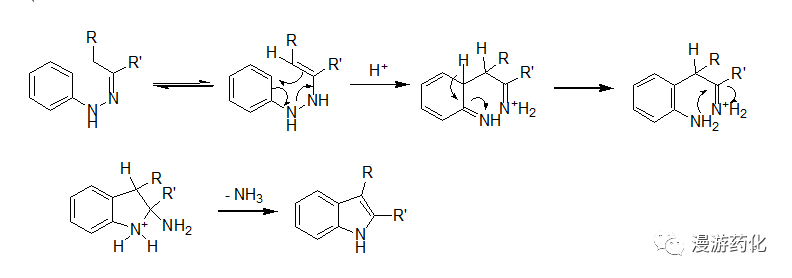
Fischer 吲哚合成法是一个常见的吲哚合成方法。通过苯腙在酸催化下加热重排消除一分子氨得到2-取代或3-取代吲哚衍生物。在实际操作中,常可以用醛或酮与等当量的苯肼在酸中加热回流得到苯腙,其在酸催化下立即进行重排、消除氨而得到吲哚化合物。常用的催化剂有氯化锌、三氟化硼、多聚磷酸等,常用的酸有AcOH, HCl, 三氟乙酸等。其机理大致如下:
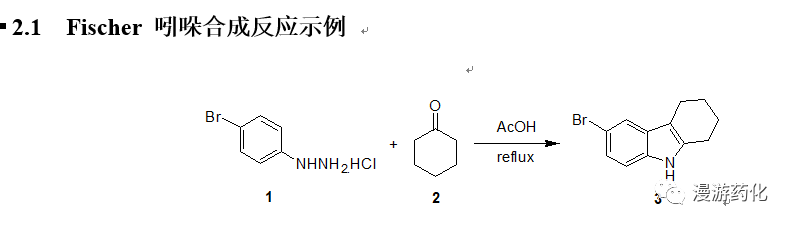
4-Bromophenylhydrazine hydrochloride 1 (21 g) was suspended in 150 mL of aceticacid, and the mixture was heated to reflux. Then a solution of cyclohexanone2 (9.3 mL) in 10 mL of aceticacid was added dropwise. After theaddition, the mixture was stirred under reflux for another 2 h. Water (50 mL) was added dropwise slowly,cooled to room temperature, the solid was filtered, washed with water, dried,pale brown solid 3 (21.65g,91 %) was obtained.
Ref: (a) B. Robinson, Chem. Rev., 1963, 373. (b) B. Robinson, Chem. Rev.,1969, 227. (c) H. Ishii, Accts. Chem. Res., 1981, 233. (d) B. Robinson, The FischerIndole Synthesis, 1982, 923. (e) D. L. Hughes, Org. Prep. Proced.Int., 1993, 607. (f) S.M. Hutchins, K. T. Chapman, Tetrahedron Letters, 1996, 4869. (g) O. Miyata et al., ibid.1999, 3601. (h) S. Wagaw et al., J. Am. Chem. Soc., 1999, 10251.
3. 从硝基苯的衍生物出发合成吲哚
对于2,3位没有取代基的吲哚,一般工业上大多采用硝基苯的衍生物出发合成,邻甲基、邻甲酰基、邻氰乙基、邻乙烯基、及邻位有氢的硝基苯衍生物都可通过相应的方法得到吲哚。

3.1 邻甲基硝基苯衍生物合成吲哚
该方法是目前最常用的,邻甲基硝基苯衍生物与DMF-DMA反应后得到相应的烯胺,然后硝基可通过多种方法还原后加成得到吲哚。还原方法一般通过加氢,但当分子内有敏感官能团(比如:Br,I都可或烯烃等)存在时可通过化学还原如:NH2NH2-RaneyNi, 铁粉,TiCl3, 锌粉还原得到吲哚。
3.1.1 邻甲基硝基苯衍生物合成吲哚示例
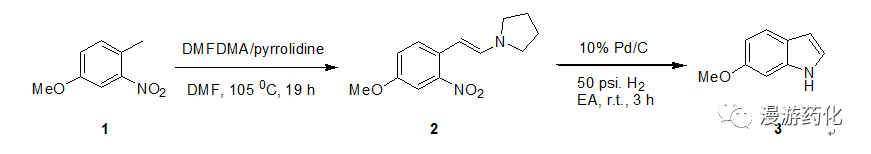
To a solution of 4-methoxy-2-nitrotoluene 1 (17.9 g, 0.107 mol) in 200 mL of dry DMFwas added DMFDMA (42 mL, 0.316 mol) and pyrrolidine (10 mL, 0.12 mmol). The mixture was heated at 105 0C for 19 h under nitrogen,then cooled, diluted with water and extracted with ether (8×50 mL). The ether layer was extracted with water (3×25 mL), dried with sodiumsulfate, and concentrated to give a deep red oil 2 which was dissolverin ethyl acetate (150 mL), and to the solution was added 10% palladium oncarbon (1.8 g). Hydrogenation at 50 p.s.i. with shaking for 3h and then filtration through celite gave a light brown filtrate. This filtrate is evaporated to purple oil,which was purified by chromatography on silica gel (eluent: DCM) to give6-methoxyindole Yield: 76%
Ref: (a) Feldman, et al, Synthesis, 1986,735. (b) Kline.T.B. et al, J.Med. Chem.,1982, 908. (c) Schumacher, R.W. et al, Tetrahedron,1999, 935. (d) bromidge, S.M.,et al, J. Med. Chem., 1998,1598. (e) Maehr, H. et al, J. Org. Chem. 1984, 1549. (f)Nicolaou, K.C. et al, J. Am. Chem. Soc., 2004, 10162.
3.2 邻甲酰基硝基苯衍生物合成吲哚
该邻甲酰基硝基苯衍生物与硝基甲烷反应后得到相应的不饱和硝化物再还原后得到吲哚。
3.1.2邻甲酰基硝基苯衍生物合成吲哚示例
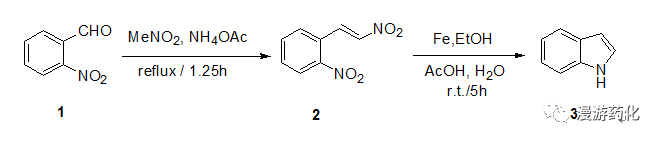
To a solution of 2-nitro-benzaldehyde 1 (3.14 g, 0.02 mol) in nitromethane (40 mL)was added ammonia acetate (0.9 g,0.012 mol) under N2 protected. Then it was heated to reflux for 1.25 h. After cooled to room temperature, it was poured into water and stirredfor 30 min. Then it was extracted withDCM (50 mL×3), and the combined organic layer was washed with brine, dried overNa2SO4 and evaporated under vacuum. The residue was purified by flash columnchromatography to yield 1.2 gpure 2-(2-nitro-vinyl)-nitrobenzene 2. Yield: 42%
To a solution of 2-(2-nitro-vinyl)-nitrobenzene 2(1.0 g, 0.005 mol) inethanol (10 mL), glacial acetate acid (10 mL) and water (3 mL) was added ironpowder (5.7 g, 0.1 mol)portionwise. After the addition, it washeated to 50 °C for 30 min. After cooled to room temperature, aq. NaHSO3was added to it and extracted with ether (50 mL×3). The combined organic layer was washed withsaturated aq. NaHCO3, dried over Na2SO4 andevaporated under vacuum. The residue waspurified by flash column chromatography to yield 0.45 g 1H-indole 3. Yield: 75%
Ref: (a) Sinhababu, Achintya K.;Borchardt, Ronald T., J. Am. Chem. Soc., 1985, 7618, (b) He,Feng; Bo, Yunxin; Altom, Jason D.; Corey, E. J.; J. Am. Chem. Soc.,1999, 6771.
3.3 邻氰甲酰基硝基苯衍生物合成吲哚示例

To a solution of 2-nitro-1-naphthyl-acetonitrile (33g, 0.155 mol) in 630 mL of ethanol containing10% water and 6.3 mL of pure acetic acid was added 19 g of 10% palladium-on-carbon. Then it was stirred at r.t. under 4 bars ofhydrogen. After the reaction completed,the catalyst was filtered and the filtration was concentrated under reduced pressure. Then residue was dissolved in 250 mL of DCM,washed with 100 mL of 0.1 N KOH solution and then dried over Na2SO4,evaporated under reduced pressure to give the crude product, which was purifiedby column chromatography using cyclohexane/EA=4:1 as eluant to yield 13 g of 3H-benzo[e]indole. Yield: 50%
Ref: (a) Makosza, M. et al., Tetrahedron, 1995,7263. (b) Bromidge, S.M. et al., J. Med. Chem., 1998, 1598.
3.4 邻乙烯基硝基苯衍生物合成吲哚示例
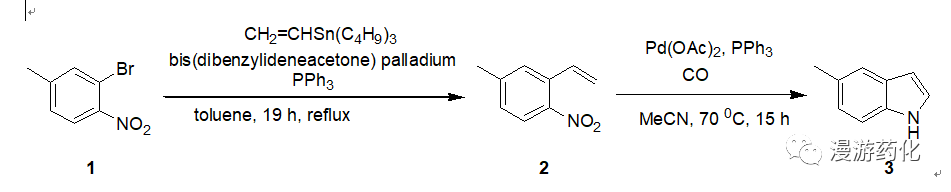
To a solution of 2-bromo-4-methylnitrobenzene 1 (1.00 g, 4.61 mmol) and vinyltri-n-butyltin(1.61 g, 5.07 mmol) intoluene (25 mL) was added, under a positive flow of argon,bis(dibenzylideneacetone) palladium (0)(265 mg, 0.46 mmol) together withtriphenylphosphine (498 mg, 1.90 mmol). Thesolution was heated at reflux (19 h) whereupon a red solution containing a blackprecipitate was formed. The reactionmixture was cooled to ambient temperature, and the solvent was removed to giveblack oil. The oil was dissolved indichloromethane (50 mL), washed with NH4OH (10%, aq, 3 x30 mL), and dried(MgSO4). Removal of solvent gave yellowoil containing a smaller amount of black viscous oil. The crude product was purified bychromatography (hexanes-EtOAc, 19:1) to give 2-ethenyl-4-methylnitrobenzene(589 mg, 3.61mmol, 78%) as yellow oil 2.
To an oven-dried, threaded ACEglass pressure tube was added 2-ethenyl-4-methylnitrobenzene 2 (152 mg,0.93 mmol), Pd(OAc)2(13 mg, 0.06mmol), triphenylphosphine (62 mg, 0.24 mmol), and 4 mL of MeCN. The tube was fitted with a pressure head, thesolution was saturated with CO (four cycles to 4 atm of CO), and the reactionmixture was heated to 70 °C(oil bath temperature) under CO (4 atm) until all starting material wasconsumed (15 h) as judged by TLC. Thereaction mixture was diluted with HCl (aq, 10%, 10 mL) and extracted with Et2O (3x10 mL). The combined organic phases were washed withHCl (aq, 10%, 10 mL) and dried (MgSO4), and the solvent was removed to give the crude product. The crude product was purified bychromatography (hexanes-EtOAc, 9:1) to give 5-methylindole 3 (62 mg,0.47 mmol, 51%) as faint yellow crystals.
Ref: Soederberg, B. et al, J.Org. Chem., 1997, 5838
3.5 邻位有氢的硝基苯衍生物直接用乙烯格氏试剂合成吲哚(Bartoli反应)示例

The 2-nitrotoluene (685 mg, 5mmol) was placed in a two–necked round bottomed flask fitted with a gas inlet(argon) and rubber septum. The flask was purged several times with argon beforeadding THF (35–40 ml) and cooling to between –40 and –45 °C. The Grignard reagent (3 eq.) was then addedrapidly in one portion to the THF solution and stirring continued for a further30 mins to 1 hour (exact length of time had little effect on yield). Saturatedammonium chloride solution was added to the reaction mixture (at ca. –40 °C) before allowing the mixture towarm to room temperature. The mixture was thoroughly extracted with diethylether (2 x 200 ml), the ether extracts combined and thoroughly washed withfurther ammonium chloride (300 ml), water (300 ml) and brine (300 ml) beforedrying (MgSO4) and concentrating in vacuo to give a dark brown gum, which was purifiedby flash column chromatography (hexane:ethyl acetate 9:1) to give 465 mg of7-methyl-indole. Yield: 71%.
Ref: (a) Adrian P. Dobbs,Martyn Voyle, Neil Whittall, Synlett, 1999, 1594, (b) Curtin, M.L et al, J.Med.Chem.,1998, 74.
4. 从苯胺的衍生物出发合成吲哚
从苯胺的衍生物合成吲哚虽不常用,但还是有一些方法被报道。
4.1苯胺经佛克烷基化再还原关环合成吲哚
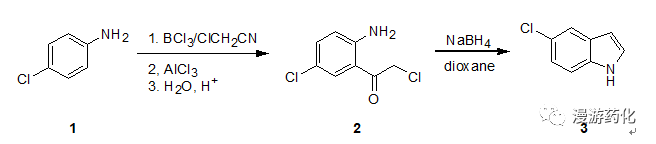
Toa stirred solution of boron trichloride (645 mg, 5.5mmol) in drybenzene (6 mL), a solution of 4-chloroaniline 1(638mg, 5 mmol) in dry benzene (6 mL) was added dropwise underice-cooling. To the resulting mixture containing 4-chloroanilineborontrichloride complex, chloroacetonitrile (0.38 mL, 6 mmol) and aluminumtrichloride (734 mg, 5.5 mmol) were added successively. The mixture was thenrefluxed for 6 h under nitrogen, becoming a solution of two layers. The evolvedhydrogen chloride was absorbed through a drying tube containing silica gel orcalcium chloride to a surface of aqueous sodium hydroxide. After cooling, ice 2N hydrochloric acid was added and a yellow precipitate was formed. To hydrolyzethe ketimine of 2 the mixturewas warmed at 80 °Cunder stirring, until the precipitate had dissolved (ca. 30 min). The cooledmixture was extracted with chloromethane (three times) and the organic layerwas washed with water, dried (MgS04), and concentrated. The neutralfraction obtained (744 mg) was recrystallized to obtain pure 2 (674 mg). Yield: 66%. The acidiclayer was made alkaline with 2 N sodium hydroxide and extracted withdichloromethane. Washing, drying, and evaporation of the solvent gave the basicfraction (170 mg). Thin-layer chromatographic purification (silica gel,chloroform containing 10% methanol) gave recovered 1 (103 mg).
To a stirred solution of 5-chloro-2-amino-α-chloroacetophenone2(204 mg, l mmol) indioxane (5mL) containing water(0.5 mL) was added sodium borohydride (1.1 mmol) and the solution was refluxedfor 5.5 h. After removal of the solvent, water was added and the mixture wasextracted with dichloromethane. The extract was dissolved in benzene and passedthrough a silica gel layer (ca. 2 g)to remove a polar fraction. The eluate with benzene was concentrated givingindole 3 (one spot, on TLC,dichloromethane). Yield: 69%
Ref: (a) T. Sugsawa, M. Adachi, K. Sasakura, A.Kitagawa, J. Org.Chem., 1979, 578, (b) Gonzalez, J.C. et al, Synthesis, 2002,475.
4.2 N-羟基苯胺DMAP催化下与丙炔酸酯缩合合成3-羧酸吲哚衍生物
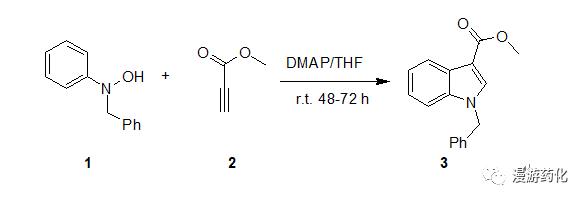
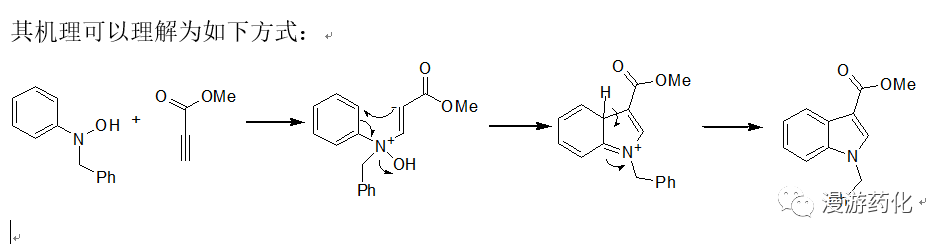
To a solution of N-benzyl-N-phenylhydroxylamine1 (86.2 mg, 0.426 mmol) in THF (15.0 mL) was added 4A molecular sieves. DMAP (6.0 mg, 0.049 mmol) and methyl propiolate 2 (54 mg, 0.562 mmol) were addedto it at 0°C. The reaction mixture was stirred at 0°C for 1 h, then at room temperature for48 h. Ethyl acetate (5.0 mL) was added, and after filtration, the organicsolution was washed with water (3 x 20 mL), brine, and then dried overmagnesium sulfate. Following filtration, the organics are concentrated underreduced pressure and the resultant oil purified by flash column chromatography(hexanes: ethyl acetate= 7:3) as eluent to give 1-benzyl-1H-indole-3-carboxylicacid methyl ester 3 (95.6 mg; 82% yield) as a white solid (m.p. 67.0-67.5°C).
Ref: (a) R.Hwu, H.V. Patel, R.J. Lin, and M.O. Gray,J. Org. Chem., 1994, 1577
4.3 Nenitzescu吲哚合成
Nenitzescu是一类比较特殊的吲哚合成方法,它的最终产物一般都是在N原子上有芳香环的化合物。对于Nenitzescu反应而言,最后一步合环反应采用不同的溶剂会得到不同的合环产物。如下的化合物4和5所示。

To a solution of (E)3-amino-but-2-enenitrile 1 (1.0 g, 12.2 mmol) in acetic acid (1.54 g, 25 mmol) and water (5 mL) wasadded aniline 2 (1.13 g,12.2 mol) at r.t.. After stirring for 30min., the mixture was cooled in an ice bath and the product 3was collected on a filter, dried invacuum. (The mixture also can beextracted with acetic ether if there was no precipitate appearance.)
To the solution of 1,4-benzo quinone (0.96 g, 9.0 mmol) in acetic acid (4 mL)was added acetic anhydride (0.8 mL) at r.t.. After stirring for 30 min., a solution of(E)3-Phenylamino-but-2-enenitrile3(1.18 g,7.5 mmol) in acetic acid (4 mL) was added to it and the mixture was stirredovernight. Crude solid was collectedafter filtered, washed with a little acetic acid and water, dried invacuum. The solid was purified by columnchromatography on silica gel using EtOAc/petro ether (1:2) as eluent to yield 6-hydroxy-3-cynao-2-methyl-1-phenyl-indole 4. (30%)
Ref: (a) R. K. Brown, The Chemistry of HeterocyclicCompounds, (b) W. J. Houlihan, Ed., 1972, 413, (c) G. R. Allen, Jr.,Org. React. 1973, 337, (d) Synthetic applications: U.Kuecklander, W. Huehnermann, Arch. Pharm. 1979, 515, (e) J. L. Bernier, J. P.,Henichart, J. Org. Chem. 1981, 4197, (f) M. Kinugawa et al.,J. Chem. Soc. Perkin Trans. I, 1995, 2677; (g) J. M. Pawlak et al., J.Org. Chem. 1996, 9055.
5. 2-叠氮基-3-芳基丙烯酸酯环合合成2-羧酸吲哚衍生物
通过叠氮基丙烯酸酯与芳香醛缩合可以得到2-叠氮基-3-芳基丙烯酸酯,其加热环合生成吲哚2-羧酸酯衍生物,一般而言只有富电子的芳环(带推电子苯环,呋喃,噻吩,吡咯)可通过该方法环合。由于反应放出氮气,在环合时一定要严格控制2-叠氮基-3-芳基丙烯酸酯滴加速度及反应瓶敞口,否则很容易喷发出来。
5.1 2-叠氮基-3-芳基丙烯酸酯环合合成2-羧酸吲哚衍生物示例

To a solution of NaN3 (60 g, 0.92 mol) in 240 mL of DMF wasadded dropwise chloro-acetic acid methyl ester 1 (75 mL, 0.86 mol) at 0 °C. After the addition, it wasallowed to warm to r.t. and overnight. The reaction mixture was poured into 1.5 L of water and extracted with ether (500mL×3). The combined organic layer waswashed with brine, dried over Na2SO4 and evaporated underreduced pressure to yield 78.4 gyellow oil azido-acetic acid methylester 2. Yield: 90%
To 500 mL of methanol was added portionwise sodium (15.7 g, 0.68 mol). After the addition, it was heated to refluxfor 30 min. Then a solution of4-methoxy-benzaldehyde 3 (46.2 g, 0.34 mol) and azido-aceticacid methyl ester 2 (78 g,0.68 g) in 100 mL ofmethanol was added dropwise to it. Afterthe addition, it was stirred at 5 °C for 2 h and overnight at r.t.. Then the reaction mixture was poured into icewater and stirred for 10 min. Deposited and filtered. The solid was washed with water (50 mL×3),dried under vacuum to yield 54.3 g yellow solid 2-azido-3-(4-methoxy-phenyl)-acrylicacid methyl ester 4. Yield: 69%
To 600 mL of xylene was slowly added dropwise asolution of compound 4 (54 g, 0.23 mol) in 400 mL of xylene atreflux. After the addition, it wasreflux for 1 h and cooled to room temperature and stirred overnight. The solvent was removed under reducedpressure and the residue was recrystallized with xylene to yield 28 g white solid 6-methoxy-1H-indole-2-carboxylic acid methyl ester 5. Yield: 59%
Reference: (a) Coowar, D. et al, J.Med. Chem., 2004, 6270, (b) Blair, J. B., et al, J. Med. Chem.,2000, 4701
关键词:吲哚 合成
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2025 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1






 发询盘
发询盘