从上世纪80年代开始,紫杉醇(taxol, 图1)由于其优异的抗癌活性吸引了学术界和工业界的极大兴趣。最初,紫杉醇是从紫杉树中分离而来,但是由于紫杉树较为稀有,所以仅仅从紫杉树里分离提取并不能满足临床上的药用需求。
为了解决这个问题,化学家发展了两种方法:第一种是利用半合成法,即通过使用一种廉价易得的前体来合成紫杉醇,这样既能获得大量紫杉醇,同时也能避免对紫杉树的大量砍伐;另一种方法是利用合成生物学手段,即通过植物细胞发酵,能够以吨级规模直接制备紫杉醇。
紫杉醇作为一个明星分子,除了具有生物活性外,其结构也极具特色。首先,紫杉醇具有在天然产物中并不常见的6-8-6三环碳骨架;其次,紫杉醇高度官能团化,不同氧化态的官能团密集分布在四个环上。正是这些原因吸引了无数合成化学家对紫杉醇的全合成研究。迄今为止,Nicolaou、Holton、Wender、Danishefsky、Kuwajima及Mukaiyama等课题组相继报道了紫杉醇的全合成。
值得一提的是,有机化学领域的传奇人物Mukaiyama教授一生致力于发展简洁高效的有机新反应,许多经典反应(如烯醇硅醚参与的Aldol反应、氧化还原诱导的脱水反应、Mukaiyama水合反应、Mukaiyama氧化反应等)都由其开发。Mukaiyama教授唯一报道的关于复杂天然产物的合成就是紫杉醇的全合成。
另外一个故事就是Robert Holton教授通过紫杉醇的相关专利获得了巨额财富,通过向药物公司转让其紫杉醇半合成的专利,Holton教授及其工作单位佛罗里达州立大学获得了超过3.5亿美元的收入。虽然从上世纪90年代初开始,陆陆续续已经有30个课题组完成了紫杉醇相关天然产物的全合成,但是仍有许多合成化学家致力于发展更独特的合成路线。此外,紫杉醇和相关的紫杉烷类组成了一个包含450多种天然产物的大家族,这些结构复杂的化合物即是制药领域的宠儿,又是合成化学家的挑战。

图1. 紫杉醇。图片来源:J. Org. Chem.
提起美国斯克利普斯研究所(The Scripps Institute)的Phil S. Baran教授,大家都不陌生,毕竟他的大名在有机化学界可以说是如雷贯耳。除了发展一系列高效的新型有机反应外,Baran教授的主要研究兴趣还是围绕天然产物的全合成,迄今为止,其课题组已经完成了许多合成难度极高的天然产物,其中包含生物碱palau’amine及甾体化合物Vinigrol的首次全合成。此外,Baran教授也提出了一系列理念,比如“氧化还原经济性(redox economy)”和“两相法(two-phase synthesis)”合成天然产物(图2)。
今天,笔者带大家解读一下Baran教授课题组发表在Journal of Organic Chemistry上的观点文章,回顾性地分析了他们历时13年的紫杉醇合成之旅,总结讨论了用“两相法”全合成紫杉烷类的战术和战略。
“两相法”合成策略的灵感源于大自然中萜类天然产物的两相生物合成过程,在酶的参与下,可以分为关环阶段(cyclase phase)和氧化阶段(oxidase phase)。而在合成化学实验室中进行的两相法全合成,与生物合成过程相比最大的挑战在于不具备选择性和催化效率极高的酶工具。在化学“两相法”合成策略中,关环阶段主要是围绕天然产物骨架的构建,即各种碳碳键的构建;而氧化阶段是在关环阶段之后,通过利用氧化反应尤其是碳氢键的直接氧化,在天然产物骨架上引入合适的氧化态,从而得到目标天然产物。
从理论上来讲,该合成策略可以避免许多重复的氧化还原过程,使得合成步数大大减少。利用该策略,Baran教授课题组已经完成了ingenol、phorbol及thapsigargin的全合成,近期该名单上又加入了紫杉醇的名字(J. Am. Chem. Soc., 2020, 142, 10526–10533)。
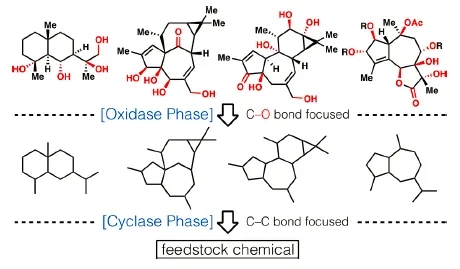
图2. 基于两相逆合成逻辑的含氧萜烯合成。图片来源:J. Org. Chem.
事实上,紫杉醇的关环阶段在2012年就已经完成了(图3,Nat. Chem., 2012, 4, 21−25)。具体而言,从廉价的原料8和9出发,通过简单的官能团转化即可获得中间体10和11。利用丁基锂把烯基溴10转化为相应的烯基锂试剂后,在铜催化剂的作用下,烯基锂试剂可以与11发生1,6-不对称加成反应而生成12。随后,通过利用三甲基铝为甲基化试剂,在铜催化剂和手性配体的作用下,中间体12可以转化为14,后者在Lewis酸催化下,通过aldol缩合反应和氧化反应的串联过程与丙烯醛反应生成中间体16。在Lewis酸作用下,16中的双烯和亲双烯体可以发生分子内Diels-Alder反应生成中间体17,该反应也标志着关环阶段的完成。最后,将17中的羰基转化为相应的烯基三氟磺酸酯后,通过利用Negishi偶联反应将其转化为关键中间体6(taxadienone)。
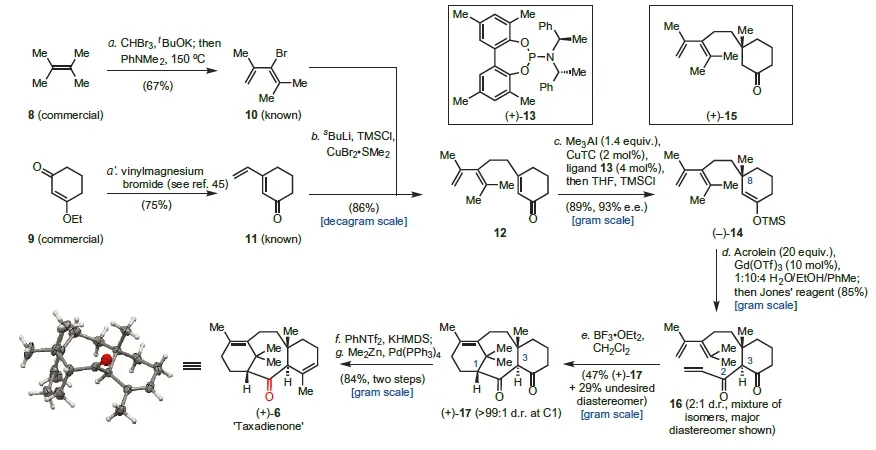
图3. 紫杉醇环系骨架的构建。图片来源:Nat. Chem.
在完成了碳环骨架构建后,作者就开始了第二个过程,即紫杉醇相应氧化态的构建。该步骤最大的挑战就是选择性问题(包含位置选择性和化学选择性),即氧化反应必须在指定的碳原子上发生,同时氧化反应需停留在特定阶段,比如有的位置需要停留在醇阶段而有的位置需要停留在羰基阶段。
除了选择性问题,不同碳原子被氧化的先后顺序对最终路线也会有决定性影响,这是因为一旦改变了分子中某个碳原子的氧化态后,整个分子的电荷分布就会发生变化,从而会改变后续氧化反应的选择性。所以,在保证选择性的同时,如何确定不同碳原子被氧化的先后顺序也是该阶段能否成功的关键。
因此,在着手紫杉醇的全合成前,作者先利用两相法合成了氧化程度较低的紫杉醇类似物taxuyunnanine D (J. Am. Chem. Soc., 2014, 136, 4909–4912)和taxabaccatin III(Angew. Chem. Int. Ed., 2016, 55, 8280-8284)。这是因为紫杉醇环系骨架含有20个碳原子,而其中8个碳原子和氧原子相连,在这样复杂的环境下通过碳氢键的氧化获得最终产物,其难度可想而知。在经过将近8年的探索后,作者终于找到了一条合适的氧化路线,在前期环系产物的基础上,通过碳氢键的氧化实现了紫杉醇的两相法全合成(图4,J. Am. Chem. Soc., 2020, 142, 10526-10533)。
正如前面所介绍的,利用简单的原料10-13,紫杉醇的环系骨架14就可以被高效地构建,后者在Cr(V)试剂16的作用下可以选择性地氧化烯丙位碳氢键而形成中间体15。随后,15在二溴化铜的作用下选择性地在C-5位进行溴化得到中间体17。由于中间体17中的双键一端已被氧化,因此在NBS及自由基引发剂的作用下,17中的双键另一侧烯丙位(C-10位)可以被溴化,并通过简单的亲核取代反应将该溴代中间体转化为TES保护的烯丙醇中间体18,后者在碱的作用下发生消除反应得到双烯酮中间体19。随后,通过甲基格式试剂对其中一个烯酮的羰基进行加成、用DIBAL对另一个烯酮的羰基进行还原以及用氘代氢化铝锂对羰基进行还原,并安装TBS保护基,将中间体19转化为20,后者在DMDO的作用下将双键氧化为环氧化物,同时将其中一个三级碳中心(C-1位)氧化为相应的醇21。接着,通过Ley氧化将21中的C-2位醇羟基氧化为羰基而得到中间体22。随后,在Na/i-PrOH的作用下将C-2位羰基又还原为醇23。需要指出的是,该氧化还原过程虽然没有改变碳原子的氧化态,但是醇的立体构型却发生了改变。被还原后的醇23随后在三光气的作用下转化为碳酸酯中间体24。接着,通过Lewis酸催化的环氧开环、碱促进的消除反应、醇保护以及DMDO氧化过程,24可以被转化为中间体27。对于中间体27的环氧基团,三价肽还原剂可以将其还原为开环的醇,在简单的保护(BOM)后而获得中间体28,后者通过Burgess消除反应便可以转化为端位烯烃中间体29。然后,将29中的C-5位羟基转化为相应的甲磺酸酯后,立即使用四氧化锇氧化剂进行双羟基化得到邻二醇30,后者在碱(DIPEA)的作用下转化为氧杂环丁烷,紧接着通过IBX氧化反应将C-10位的OTES直接氧化为中间体31。以31为原料,通过KOt-Bu/(PhSeO)2O将31中羰基的邻位(C-9位)氧化为羟基而生成中间体32,后者通过强碱诱导的氢转移过程将32中的羰基和羟基发生互变,通过进一步的Ac保护转化为中间体33。最后,通过脱保护和侧链安装步骤,成功地实现了紫杉醇的氧化阶段,这也标志着两相法合成紫杉醇的大功告成(图4)。

图4. 紫杉醇合成的氧化阶段。图片来源:J. Am. Chem. Soc.
总结
在这篇文章中,Baran课题组系统地总结了他们在两相法合成紫杉醇的氧化阶段所做的各种探索,特别是在此期间他们所尝试的各种碳氢键氧化方法对笔者颇具启发。虽然他们发展的方法对于紫杉醇本身的药物活性研究帮助并不大,但是他们从关环阶段到氧化阶段获得的各种具有不同氧化态的中间体却有可能具有重要的价值。由于从最低氧化态到最高氧化态的合成中会得到许多天然产物的类似物,如果天然产物具有生理活性,可以合理预测这些天然产物类似物也有可能具有一定程度的生理活性(尽管活性会低于天然产物)。
从这个角度来讲,与传统的天然产物合成策略“聚合法”(convergent route)仅能得到天然产物本身相比,利用“两相法”来合成天然产物就具有特殊的意义,对于紫杉烷家族以及其他萜烯家族天然产物的合成也有指导意义。
关键词:两相法 合成 紫杉醇 中间体
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2025 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1






 发询盘
发询盘